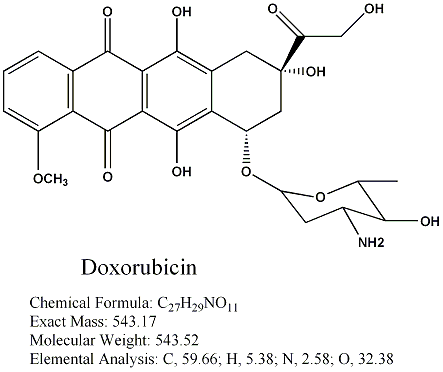
Systematic (IUPAC) Name
(8S,10S)-10-(4-amino-5-hydroxy-6-methyl-tetrahydro-2H-pyran-2-yloxy)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)
-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione
CAS number 23214-92-8
PubChem 31703
DrugBank APRD00185
Introduction
Doxorubicin(DXR) is an anthracycline antibiotic used in cancer chemotherapy to treat a wide variety of malignancies. It is also called 14-OH-daunorubicin, Adriamycin and Doxil, which is a liposome encapsulated version. Daunorubicin, another anthracycline antibiotic, is the immediate precursor of DXR in its biosynthetic pathway, and is more abundantly found as a natural product because it is produced by a number of different wild type strains of bacteria. In contrast, only one known non wild-type species, streptomyces peucetius subspecies cesius ATCC 27952, was initially thought capable of producing the more widely used doxorubicin [1]. This strain was discovered by Arcamone et. al [2] in 1969 by mutating a non-doxorubicin producing species of streptomyces peucetius that was able to produce daunorubicin. Subsequently Hutchinson's group has shown that creating special environmental conditions, or by the introduction of genetic modifications, other strains of streptomyces can produce doxorubicin [3]. His group has also cloned many of the genes required for DXR production, although not all of them have been fully characterized. In 1996, Strohl's group discovered, isolated and characterized, the gene that converts daunorubicin into DXR and named it dox A [4]. By 1999 they produced recombinant Dox A, a cytochrome P-450 oxidase, and found that it catalyzed multiple steps in the DXR pathway, including steps leading up to daunorubicin [5] (see Figure 3.). This was significant because it became clear that all daunorubicin producing strains must posess the necessary genes to produce doxorubicin, the much more therapeutically important of the two. Hutchinson's group went on to develop methods to improve the yield of DXR, from the fermentation process used in its commercial production, not only by introducing plasmids causing increased expression of Dox A, but also by mutating genes in streptomyces peucetius that encode enzymes that shunt DXR precursors to less useful products, for example baumycin-like glycosides [1]. Some triple mutants, that also overexpressed Dox A, were able to double the production of DXR. This is of more than academic interest because at that time DXR cost about $1.37 million per kg and current production in 1999 was 225 kg per annum [6]. More efficient production techniques have brought the price down to $1.1 million per kg for the non-liposomal formulation (AWC). Although DXR is produced semi-synthetically from daunorubicin, the process involves electrophilic bromination and multiple steps [7] and the yield is poor. Since daunorubicin is produced by fermentation anyway, it would be ideal if the bacteria could complete the process more effectively.
Mechanism of Action
DXR and the other anthracyclines are classic DNA intercalating agents that seem to have their most profound effects during cell replication, and to a much lesser extent during transcription [8]. The primary mechanism of action is not completely clear because these drugs effect a myriad of changes, but it is likely related to their interaction with topoisomerase II causing DNA strand breaks during DNA replication leading to cell cycle arrest, cell death and apoptosis. DXR is also able to induce interstrand DNA cross linking in both nuclear and mitochondrial DNA, and some investigors believe the cross linking capablility of the anthracylines is more closely correlated with their potency than is their Topo II inhibition [9]. DXR is also able to produce intracellular free radicals which contribute to its cytotoxicity by enhancing its ability to form adducts with DNA and intracelular proteins [10]. DXR can be reduced to a semiquinone free radical that can react with molecular oxygen to yield additional free radicals: superoxide and hydroxyl. Iron must be present for these reactions to result in significant damage. Doxorubicin binds to iron and the iron-doxorubicin complex catalyzes the free radical reactions. Much research is being done to develop new drugs in the anthracycline class that have higher therapeutic indices and also to find other drugs that reduce or reverse the toxic effects without interfering with the clinically relevant actions [11].
Clinical Use
Doxorubicin (DXR) is almost always used in combination with other chemotherapeutic agents. It remains a mainstay in the treatment of some malignancies, while it is used for salvage therapy in others. DXR has been used successfully to produce regression in disseminated neoplastic conditions such as acute lymphoblastic leukemia, acute myeloblastic leukemia, Wilms’ tumor, neuroblastoma, soft tissue and bone sarcomas, breast carcinoma, ovarian carcinoma, transitional cell bladder carcinoma, thyroid carcinoma, gastric carcinoma, Hodgkin’s disease, malignant lymphoma and bronchogenic carcinoma in which the small cell histologic type is the most responsive. Doxorubicin is also indicated for use as a component of adjuvant therapy in women with evidence of axillary lymph node involvement following resection of primary breast cancer [12]. In contrast, the more easily obtainable daunorubicin is only used for two types of leukemias.
Unfortunately, DXR administration is limited by cumulative dose-dependent cardiotoxicity, which is only somewhat reduced by using the much more expensive liposomal formulation. To decrease the risk of myocardial toxicity, heart function must be measured at baseline and then monitored regularly during treatment, particularly if a high cumulative dose is anticipated. Echocardiography, radionucleide scans and even sometimes heart biopsies are used to guide therapy if DXR is felt to be essential in patients with underlying heart disease or possible myocardial toxicity [13]. Irreversible cardiomyopathy with serious congestive heart failure is a significant risk in patients who have received doses in excess of 500-550 mg/m2 of DXR. Despite its toxicity it remains one of the most effective and broadly active anti-tumour agents. Other toxicities include myelosuppression, gastrointestinal toxicity manifesting as nausea and vomiting and mucositis, and if accidenentally locally infiltrated, tissue necrosis that can be severe enough to require skin grafting.
Dexrazoxane, Zinecard [14], has recently been approved to be used to decrease the risk of cardiac toxicity. The mechanism by which ZINECARD exerts its cardioprotective activity is not fully understood. It is a cyclic derivative of EDTA that readily penetrates cell membranes. Results of laboratory studies suggest that dexrazoxane is converted intracellularly to a ring-opened chelating agent that interferes with iron-mediated free radical generation thought to be responsible, in part, for anthracycline-induced cardiomyopathy. It is approved only for use in metastatic breast cancer after 300mg/m2 of DXR has been administered. It is possible that it may decrease the anti-tumour effect of DXR by interfering with its action. Other drugs are being developed.
Biosynthesis of Doxorubicin (Overview)
The anthracycline skeleton of doxorubicin (DXR) is produced by a Type II polyketide synthase (PKS) in streptomyces peucetius. First, a 21 carbon decaketide chain is synthesized from a single 3-carbon propionyl group, derived from propionyl CoA, and 9 2-carbon units derived from 9 sequential (iterative) decarboxylative condensations of malonyl CoA (see Fig 1 and 2b). Each malonyl CoA unit contributes a two carbon ketide unit to the growing polyketide chain. Each iterative addition is catalyzed by the "minimal PKS" consisting of an acyl carrier protein (ACP), a ketosynthase (KS)/chain length factor (CLF) heterodimer and a malonyl:ACP acyltransferase (MAT).
This process is very similar to fatty acid synthesis, by fatty acid synthases (FAS) and to Type I polyketide synthesis. But, in contrast to fatty acid synthesis, the keto groups of the growing polyketide chain are not modified during chain elongation and they are not usually fully reduced. In contrast to Type I PKS systems, the synthetic enzymes (KS, CLF, ACP and AT) are not attached covalently to eachother, and may not even remain associated during each step of the polyketide chain synthesis. Also unlike Type I PKS, the growing Type II polyketide chain is not modified by reduction of the keto groups while the chain is being made, although some of the groups are modified later. The Type I "megasynthases" consist of redundant enzymes covalently linked together into a number of modules. Each module has its own AT, ACP, KS and modifying enzymes, and each is responsible for the addition of one unit to the growing chain, and the units are not always two carbons derived from malonyl CoA. The length of the chain is dependent on the number of modules in the megasynthase so no CLF is needed. [15]
Since the Type II systems are iterative, a CLF is necessary to specify chain length to ensure the desired product is produced and the chain synthesis does not continue. Since the chain is not modified during its production, and since the methylene groups of the usually longs flexible chains are relatively acidic, they react readily with the carbonyl groups in intramolecular aldol condensations to form cyclic ring systems which spontaneously, or with the help of aromatase enzymes (ARO), become aromatic. For this reason, sometimes polyketides derived from Type II systems are called aromatic polyketides.
After the 21 carbon decaketide chain of DXR is completed, successive modifications are made and eventually a tetracyclic anthracycline aglycone is the result (Fig 2.) [16]. The daunosamine amino sugar, activated by addition of TDP, is created in another series of reactions (Fig 4.) [17]. It is joined to the anthracycline aglycone and further modifications are done to produce first daunorubicin then DXR (Fig 3.) [18]. Each of these steps will be discussed.
Streptomyces, part of the gram-positive actinomycetes family of filamentous soil dwelling bacteria, produce a huge number of useful natural products by "secondary metabolism". Primary metabolism includes all things necessary for basic survival including the production of fats, sugars and amino acids as well as structural proteins, RNA etc. Secondary metabolism produces everything else, including chemoattractants, colorings, antibiotics such as DXR, that give the organism a selective advantage. These sustances are frequently toxic to other organisms and even to the producing organism, so that methods have been developed for self-protection. For example, DXR producers have evolved a set of "self resistance" genes to produce membrane pumps cause efflux of the DXR out of the cell into the environment (drr loci) [19]. Since these complex molecules are only needed in some conditions and require a lot of energy to produce, their synthesis is tightly regulated. Not only is DXR production regulated by expression its synthetic machinery, but also by the degree that its precursors are shunted away from the final DXR product [20].
There are at least 3 gene clusters important to DXR synthesis: dps genes which specifiy the enzymes required for the polyketide chain synthesis and its first cyclizations, the dnr cluster is responsible for the remaining modifications of the anthracycline structure and the dnm genes involved in the aminosugar, daunosamine, synthesis. For each step of the biosynthesis the gene products will be defined and correlated with their function. For example Dps G (the dps G gene product) is the acyl carrier protein (ACP).
Initiation of Polyketide Chain by Primer Selection (Figure 1 and 2a.)
The initial event in DXR synthesis is the selection of the propionyl CoA starter unit and its decarboxylative addition to a two carbon ketide unit, derived from malonyl CoA (via Claisen condensation) to form the five carbon B-ketovaleryl ACP. The five carbon chain is delivered by the ACP to the cysteine sulfhydryl group at the KS active site, by thioester exchange, and the ACP is released from the chain. The free ACP picks up another malonate from malonyl CoA, also by thioester exchange, with release of the CoA. (see Fig 2a).
The ACP brings the new malonate to the active site of the KS where is it decarboxylated, possibly with the help of the CLF subunit, and joined to produce a 7 carbon polyketide now anchored to the ACP. Again the ACP hands the chain off to the KS subunit and the process is repeated iteratively until the decaketide is finished.
In most Type II systems the initiating event is delivery by ACP of an acetate unit, derived from acetly CoA, to the active site of the ketosynthase (KS) subunit of the KS/CLF heterodimer, although sometimes malonate is decarboxylated to produce the initial acetate. The default mode for Type II PKS systems is the incorporation of acetate as the primer unit, and that holds true for the DXR "minimal PKS". In other words the action of KS/CLF/ACP from this system (Dps A, B and G) will not produce 21 carbon decaketides, but 20 carbon decaketides because acetate is the preferred starter. The process of specifying propionate is not completely understood, but it is clear that it depends on an additional protein, Dps C, and possibly also Dps D [21][22]. There is experimental evidence that this may occur in a number of ways:
1) The amino acid sequences of Dps C and Dps D are very similar to a fatty acid KS (KS III) and to to an acyltransferase (MAT) found in E. coli. KS III proteins start fatty acid synthesis by condensing the first acetate unit. By homology it was believed that these proteins acted together to form an "initiation module" (see Figure 1. - possibility 1) and there is some evidence that this can occur. The Dps C (KS III activity) would catalyze the first condensation to get the 5 carbon B-ketovaleryl ACP, which would then be delivered to the KS active site of KS/CLF by ACP, where 8 additional rounds of decarboxylative condensation would produce the 21 carbon decaketide. Dps D could play an as yet unidentified role, but since its absence does not seem to affect DXR prodution [6] it may only be important under unusual conditions, for example pH extremes, high levels of acetly CoA or low levels of propionyl CoA.
Some bacteria that initiate synthesis of polyketides with non-acetate primers have evolved two sets of KS, ACP and AT - one for the initial condensation and the second for subsequent chain elongations. The DXR synthetic system does not have a second ACP, although the usual ACP (Dps G) is compatible with the KS/CLF (Dps A/B) and the KS III ( Dps C) [23] so "possibility 1" in Figure 1. is a good bet. This would require that the active site of KS III (Dps C) be propionylated and then condensation could occur when ACP delivered a malonate. Some argue that the active site of Dps C does not have the very conserved cysteine thought necessary to accept a propionyl group. But it does have a serine in the same location, which has been found to be be propionylated under some conditions in vitro, and could serve as an anchor for the starter group via an ester instead of thioester linkage.
2) There is also good evidence for possiblity 2 (Figure 1.) where Dps C acts as a propionyl transferase transfering the requisite propionyl group to ACP for delivery to the KS/CLF complex where it undergoes 9 iterative decarboxylative condensations to get the desired decaketide. Dps C does have acyltransferase activity as well as KS activity, and investigators still debate the importance of each pathway[24], but both pathways probably do contribute to varying degrees depending on environmental conditions.
3) What about the role of malonyl CoA:ACP transferase (MAT)? In most Type II PKS systems a dedicated MAT has been found to be dispensible for polyketide production under in vitro conditions [25]. Plasmids containing all the enzymes necessary for a given polyketide to be produced, excluding the MAT, when expressed in heterologous hosts support polyketide production. Initially this was rationalized by hypothesizing that the PKS "borrowed" the MAT used in the heterologous host's FAS system. This certainly does occur and it may be the primary way ACP is malonylated in vivo. A recent publication [26] provides excellent evidence that "self-malonylation" is an inherent characteristic of Type II ACPs. In summary, a given Type II PKS may provide its own MAT (s), it may borrow one from FAS, or its ACP may self malonylate to accomplish the goal of producing the polyketide.

Biosynthesis of the DXR Decaketide (Figures 1 and 2a.)
After DpsC aided primer selection and initial condensation the DXR PKS acts like most other Type II PKS systems. The growing chain is anchored to the ACP, but it is transferred to the active site of the KS portion of the KS/CLF heterodimer to allow reloading of the ACP with another malonyl group. An MAT usually catlalyzes the condensation of the malonyl CoA with ACP, which releases the CoA. This is a thioester transfer from one sulfhydryl group to another. There is no absolute requirement that the malonyl ACP returning to the KS/CLF active site is the same one that was there before handing off the growing chain. Recently, using a yeast two-hybrid system to explore protein interactions in this dps system, positive interactions were not found between ACP (Dps G) and KS/ACP (Dps A/B) [27]. This implies that the interaction between ACP and the KS/CLF is transient and that it does not remain bound as an integral structural component of the complex. Rather the ACP may only form a transient interaction to sustain acyl-transfer during polyketide biosynthesis.
After DpsC aided primer selection and initial condensation the DXR PKS acts like most other Type II PKS systems. The growing chain is anchored to the ACP pantetheine arm until it is transferred to the active site of the KS portion of the KS/CLF heterodimer thereby allowing reloading of the ACP with another malonyl group. An MAT usually catlalyzes the condensation of the malonyl CoA with ACP, which releases the CoA. This is a thioester transfer from one pantetheine sulfhydryl group to another.
There is no absolute requirement that the malonyl ACP returning to the KS/CLF active site is the same one that just handed off the growing chain. Recently, using a yeast two-hybrid system to explore protein interactions in this dps system, positive interactions were not found between ACP (Dps G) and KS/ACP (Dps A/B) [27]. This implies that the interaction between ACP and the KS/CLF is transient and that it does not remain bound as an integral structural component of the complex. Rather the ACP may only form a transient interaction to sustain acyl-transfer during polyketide biosynthesis. It is unknown whether the same KS/CLF/ACP ternary complex chaperones the growth of a full length polyketide chain through the entire catalytic cycle, or whether the complex dissociates after each condensation reaction [28]. But recently a 2.0-Å resolution structure of the actinorhodin KS/CLF, which is very similar to the dps KS/CLF, was published [29]. It shows polyketides being elongated inside an amphipathic tunnel formed at the interface of the KS and CLF subunits. The tunnel is about 17-Å long and one side has many charged residues which appear to be stabilizing the carbonyl groups of the chain, while the other side is hydrophobic. This structure explains a number of things including why both subunits are necessary for chain elongation, and how the reactive growing chain is protected from random reactions until it is positioned properly for orderly cyclization. The extremely reactive chain will spontaneously cyclize without an invironment in which intramolecular reactions are prevented, and the tunnel provides a way to keep the chain extended during extension.
The structure also suggests a mechanism for chain length regulation. Amino acid side groups extend into the tunnel and act as "gates". A couple of particularly bulky residues may be impassable by the chain causing termination of its elongation. Modifications to tunnel residues based on this structure were able to change the chain length in the predetermined direction (ie: longer or shorter)[30]. Evidence was also provided that when the the final condensation occurs that the chain "buckles" allowing an intramolecular attack by the C-12 methylene carbanion, generated by enzyme catalyzed removal and stabilized by electrostatic interactions in the tunnel, on the C-7 carbonyl (see 1 in Figure 2b. for numbering). This tunnel aided intramolecular aldol condensation provides the first cyclization event and it happens when the chain is still in the tunnel. The same C-7/C-12 attack occurs in the biosynthesis of DXR, likely in a similar fashion (Fig 2 b.).
Biosynthesis (Conversion to 12-deoxyalkalonic acid)
The 21 carbon decaketide is converted to 12-deoxyalkalonic acid (3), the first free easily isolated intermediate in DXR biosynthesis in 3 steps. These steps are catalyzed by the final 3 enzymes in the dps gene cluster (Fig 2b.) and are considered part of the polyketide synthase.
While the decaketide is still associated with the KS/CLF heterodimer the 9-carbonyl group is reduced by Dps E, the 9-ketoreductase, using NADPH as the reducing agent/hydride donor. Dps F, the “1st ring cyclase” /aromatase, is very specific and is in the family of C-7/C-12 cyclases that require an antecedent reduction [31] for their action. If Dps F is coupled, to other PKS systems, it can cyclize only polyketides greater in length than octaketides, with a 9-OH group, and only at C-7/C-12. In contrast Dps E KR will only reduce decaketides or greater.
These two reactions are felt to occur while the polyketide chain is still partially in the KS/CLF tunnel or at least associated with the “minimal PKS”. It is not known whether ACP is involved or what finally cleaves the chain from its covalent link to KS or ACP. If the Dps F cyclase is inactivated by mutations or deletions, the chain will cyclize in random fashion. So Dps F is thought to “chaperone” or help fold the polyketide and protect it from random events. It is energetically favorable for the cylization, subsequent dehydration and resultant aromatization.[32]
Next, Dps Y also acts as a “chaperone” by helping further fold the molecule to regiospecifically promote the next two C-C bonds to form, and then catalyzes dehydration leading to aromatization of one of the rings and giving 3.
Biosynthesis (conversion to є-rhodomycinone)
The next set of reactions are catalyzed by enzymes originating from the dnr gene cluster, which also encodes the regulatory enzymes responsible for regulating DXR biosynthesis.
Dnr G is a C-12 oxygenase (the numbering system is not the same - please refer to Fig 2b. 3 and 4); it introduces a keto group using molecular oxygen. It is an "anthrone type oxygenase", classified as a an internal monoxygenase. These quinone-forming monooxygenases which are important 'tailoring enzymes' in the biosynthesis of several types of aromatic polyketide antibiotics have no cofactors: no flavins, metals or energy sources. Their mechanism is poorly understood but may involve a "protein radical" [33].
Alkalonic acid, the quinone, is formed as the product. Dnr C, alkalonic acid-O-methyltransferase methylates the carboxylic acid end of the molecule forming an ester, using S-adenosyl methionine (SAM) as the co-factor/methyl group donor. The product is alkalonic acid methyl ester (AAME), and also S-adenosyl homocysteine. Interestingly, this methyl group is removed later in the pathway to DXR. This methylation activates the methylene group adjacent to the ester for the next step: attack of the methylene group causing the fourth ring cyclization.
Dnr D, the fourth ring cyclase(AAME cyclase), catalyzes an intramolecular aldol addition reaction. No cofactors are requred and neither aromatization nor dehydration occur. A simple base catalyzed mechanism is proposed which does not allow the enzyme to be classified as a Type I or II aldolase [34]. The product is aklaviketone.
Dnr H, aklaviketone reductase, stereospecifically reduces the 17-keto group of the new fourth ring to a 17-OH group. This introduces a new chiral center and NADPH is used as the reducing agent/hydride cofactor.
Dnr F, aklavinone-11-hydroxylase, is a FAD monooxygenase that uses NADPH as a cofactor. The NADPH activates molecular oxygen for subsequent hydroxylation. є-rhodomycinone is the final product (Fig 2b. and 3)[35].
Biosynthesis (conversion to doxorubicin)
Dnr S, catalyzes the addition of L-daunosamine-TDP (biosynthesis in next section), to є-rhodomycinone to give rhodomycin D. The TDP released in the reaction provides the driving force. Daunosamine is 2,3,6-trideoxy-3-aminohexose, an amino sugar. The enzyme has sequence similarity to glycosyltransferases of the "unusual sugars" added to Type II PKS aromatic products
[36].
Dnr P, rhodomycin D methylesterase, removes the methyl group added a few steps previously by DnrC. It initally served to activate the adjacent metyhlene group for fourth ring cyclization, and after that it has served to protect the carboxylic acid group to which it is attached from departing from the C-10 carbon (see Fig 3). Had the carboxylic acid not been protected prior to the fourth ring cyclization, after the cyclization event its departure and the subsequent dehydration of the fourth ring would have been driven forward by the production a bicyclic aromatic ring system. After C-7 has been reduced and then glycosylated by the amino sugar, the C-8 methylene group is no longer activated for deprotonation (not made more acidic by an adjacent keto group) making aromatization more difficult
[34]. Note that the non-isolable intermediate, with numbering, is shown below in the 3rd molecule in Fig 3.(the numbering system is very odd and a vestige of early nomenclature). The decarboxylation of the intermediate occurs spontaneously or under the influence of Dnr P giving 13-deoxycarminomycin.
A crystal structure, with bound products, of aclacinomycin methylesterase, an enzyme with 53% sequence homology to Dnr P, from ''streptomyces purpurascens'' , and able to catalyze the same reaction, has been solved
[37] . It is able to catalyze the same reaction and uses a classic Ser-His-Asp catalytic triad with serine acting as the nuccleophile and gly-met providing stabilization of the transition state by forming an "oxyanion hole". The active site amino acids are almost entirely the same as Dnr P and the mechanism is almost certainly identical.
Although Dox A is the next enzyme in the synthetic scheme shown in Figure 3, Dnr K will be discussed next. Dnr K, carminomycin 4-O-methyltransferase, is shown methylating carminomycin because that was the reaction for which it was named. Actually it is able to O-methylate the 4-hydroxy group of any of the glycosylated moeities in Figure 3. prior to daunorubicin. A 2.35 A resolution crystal structure of the enzyme with bound products has recently been solved. The active site contains the O-methylated product of є-rhodomycin, 4-methoxy-є-rhodomycin, and S-adenosyl homocysteine, the result of S-adenosyl homocysteine donating its methyl group. The orientation of the bound ligands are consistent with a SN2 mechanism of methyl transfer. Site directed mutagenesis of the potential acid/base residues in the active site did not affect catalysis leading to the conclusion that Dnr K most likely acts as an entropic enzyme in that rate enhancement is mainly due to orientational and proximity effects
[38]. This is incontrast to most other O-methyltransferases where acid/base catalysis has been demonstrated to be an essential contribution to rate enhancement.
Dox A catalyzes three successive oxidations in DXR producing strains of streptomyces peucetius. The reason why other daunorubicin producers are deficient in DXR production is not primarily because of low levels or malfunctioning Dox A, but because there are many products shunted away from the pathway shown in Figure 3. Each of the glycosylated moieties is a potential target of other enzymes, not shown, some of which are products of the dnr gene cluster. Mutations inactivating some of these important shunt enzymes does significantly boost DXR production
[1] . In addition, Dox A has a very low kcat/Km value for C-14 oxidation (130/M) compared to C-13 oxidation (up to 22,000/M for some substrates). Genetic manipulation to boost Dox A production has also increased yields, particularly if the genes for the shunt enzymes are inactivated simultaneously.
Dox A is a cytochrome P-450 monooxygenase that has broad sustrate specificity, catalyzing antracycline hydroxylation at C-13 and C-14 (see Fig. 3). The enzyme has an absolute requirement for molecular oxygen and NADPH [5]. Initially, two successive oxidations are done at C-13, followed by a single oxidation of C-14 that converts daunorubicin to doxorubicin.


Biosynthesis of TDP-L-daunosamine
Only some of the enzymes involved in production TDP-L-daunosamine (TLD) have been characterized. Although the sequence of the biosynthesis as shown in Figure 4 (adapted from[39] [6]).
Glucose-1-phosphate, from primary metabolism, is converted to TDP-d-glucose in step 1. The TDP moeity remains until the sugar is fully synthesized and joined to the anthracycline aglycone. The release of pyrophospate drives the reaction forward.
The conversion of 2 to 4 occurs via an interesting mechanism involving NAD+ oxidizing C-4 of 2, which acivates the adjacent C-5 methylene group by increasing its acidity, thereby allowing removal of a proton from C-5. This resullts in a transient double bond between C-5 and C-6 (not shown) with departure of the C-6 hyroxyl group, as H20. Then a hydride is donated by NADH at C-6, reducing the transient alkene to a C-6 methyl group and giving 4. [40].
The conversion of 4 to 8 is not completely understood. It has been determned that when a gene cassette containing the dnmLMJVUTS genes, plus essential regulatory and resistance genes, is introduced into a heterologous host that is not able to make TLP naturally, it enables it to produce TLP. When any of these genes are left out of the cassette production of TLP does not occur. Thus these genes are considered the "minimal" genes necessary for the production of the TLP and its transfer to anthracyline aglycones [39]. This of course does not rule out the possibility that the host produced genes that are able to complement crucial elements missing from the gene cluster. Insertion of plasmids containing the genes sufficient for TLD synthesis and transfer, and also many other glycosides, have been introduced into heterologous host bacteria allowing the production of new products that have the potiential to be medically useful.
Johnnny is cool [50].